周敏 Nature communications文献解读

全文速览:
电催化NO3−合成氨(NH3)是一种有效的减少污染物的同时实现变废为宝的技术。然而,该过程受到多个竞争反应和NO3−阴极表面难吸附的限制。文献报道了一种Fe/Cu双原子锚定在多孔氮掺杂石墨烯的催化剂表现出高的催化活性和选择性。该催化剂可使NH3的最高法拉第效率达到92.51% (- 0.3 V vs RHE),NH3产率高达1.08 mmol h−1mg−1。计算和理论分析揭示了NO3−和Fe/Cu之间的相对强相互作用促进了NO3−阴离子的吸附及电子的释放。由于双原子位点的存在降低了整个反应的势垒,N-O键被成功削弱,从而降低了整个反应的能垒。本研究的双位点和杂原子策略为进一步的催化剂开发提供了灵活的设计,并扩展了硝酸还原和氨合成的电催化技术。
背景速览
氨是农业、塑料、制药等行业常用的重要化学品。Harber-Bosch工艺作为工业主要制备NH3的工艺,然而该工艺消耗了大量的资源和能源,并且产生了1%的二氧化碳排放,造成恶劣的环境影响。因此,清洁高效的合成氨技术日益受到人们的重视。硝态氮(NO3−)浓度的增加在地表水和地下含水层污染水资源,对人类健康构成严重威胁。NO3−是可溶且热力学稳定,从水中提取去除被认为是一项具有挑战性和长期的任务。NO3−直接转化为NH3可以减少环境污染,同时节约能源,实现可持续制氨。
单原子催化剂(SACs)由于其原子设计方便、构效关系简单、原子利用率高等优点,近年来成为催化科学研究的一个新领域。可调的局部配位和明确的活性位点使SACs成为研究NO3−RR催化剂构效关系的理想平台。通过设计活性单原子的配位环境,不同质子-电子转移步骤的能垒可以选择性地改变和降低,为提高NO3−RR的选择性提供了机会。据报道,Fe-SAC具有高活性的活性和选择性。然而,对于多电子转移反应,一个特定的位点使得SAC难以打破催化剂与中间体之间多个相似的的线性比例关系。优化速率决定步骤(r.d.s)中某一关键中间体的相互作用可将另一步骤转化为r.d.s。然而,双基点催化剂的精确设计和制造仍是一项挑战,而且杂原子基点的催化机理难以捉摸。
在这项工作中,文献提出了一种用于高效NO3-RR的双原子催化剂。活性金属原子(Fe/Cu)锚定在掺氮石墨烯(HNG)的孔边缘位点上,形成铁原子和铜原子直接成键,并且一个金属原子与两个氮原子配位的 "Y 型"ML3 配位。Fe/Cu-HNG 具有很高的活性(- 0.3 V vs RHE)时,电流密度为~ 38.5 mA cm-2和FE为92.51%。通过结合在线差分电化学质谱法(DEMS)和密度泛函理论(DFT)计算,文献揭示了从NO3−到NH3的反应途径和转化机制。对电子结构的深入分析表明,NO3−与双金属原子d轨道之间的强耦合降低了第一个阴离子吸附步骤的能垒,也就是在高电流密度下的 r.d.s。双原子异质结构可进一步削弱N-O 键,从而降低NH3生产的低能量障碍。双原子的协同效应为设计 NO3−RR催化剂的另一种设计方法。
图文解析
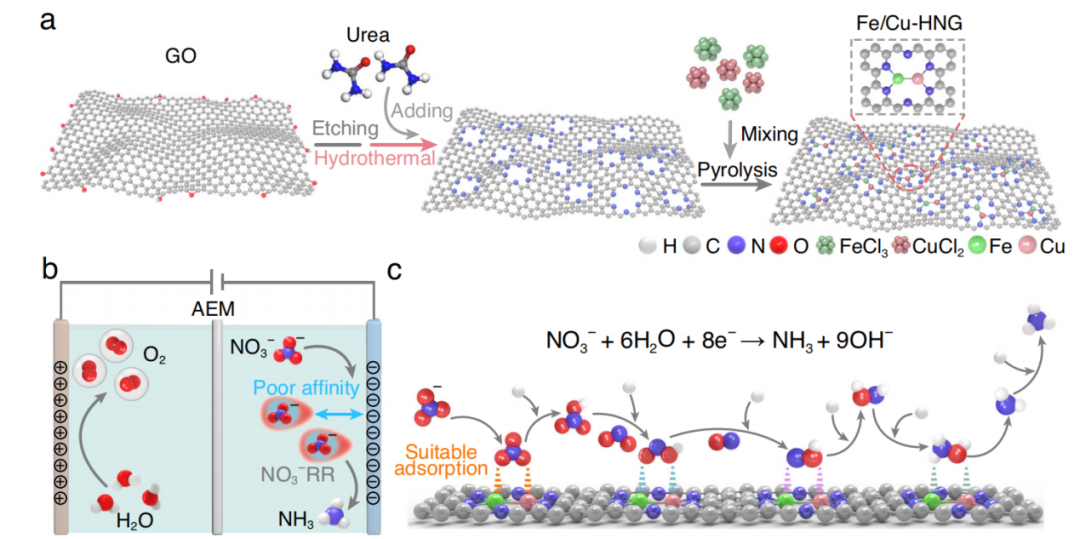
图一:Fe/Cu-HNG的结构表征
为了固定双原子并形成金属-金属二聚体(如图1a所示),本文首先在石墨烯中设计了空穴,以产生大量的边缘位点,这些边缘位点被进一步硝化以结合Fe/Cu原子。
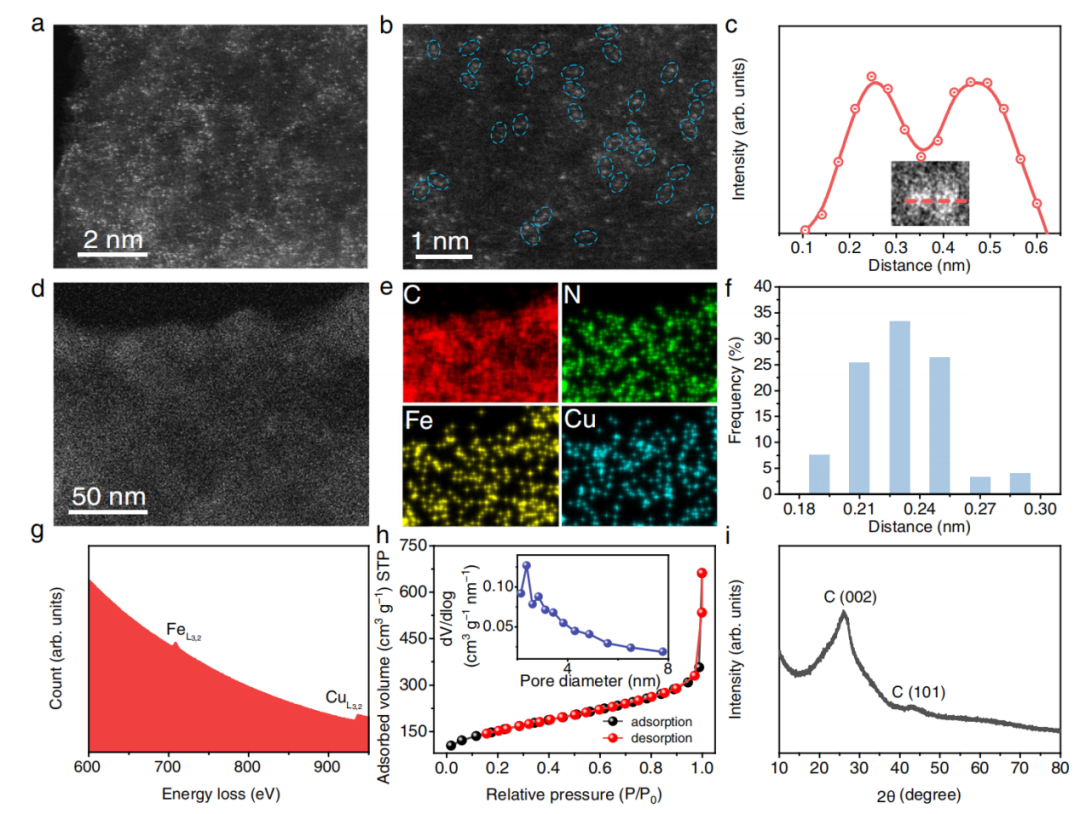
图二:催化剂结构表征
HAADF-STEM图像可以观察到大多数双原子对。EDX线扫描测出原子对(金属-金属对长度)之间距离为~2.3 Å。EDX显示了C、N、Fe和Cu的均匀分布。图2g中的EELS显示了Fe和Cu元素的共存。N2吸附-解吸等温线证实了高介孔结构的存在。XRD图显示只有堆叠石墨烯层的两个宽峰。因此得出结论,Fe/Cu双原子结构在HNG上成功构建,没有观察到金属的聚集或纳米颗粒。
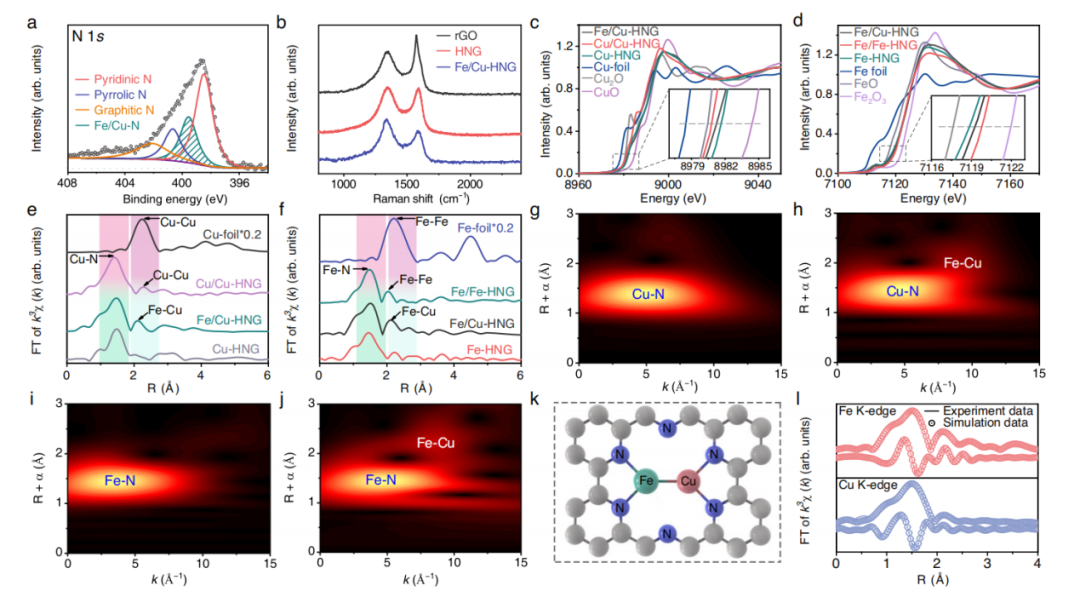
图三:催化剂结构表征
N 1s 信号的宽峰可以分为吡啶N、吡咯烷N、石墨N和Fe-N/Cu-N。拉曼光谱表明掺杂Fe/Cu会直接碳纳米片的缺陷。Cu K-edge XANES证实了Cu 的氧化态介于+1和+2之间。同样,Fe K-edge XANES位于 Fe (0) 箔和 Fe2O3(3+) 之间,表明 Fe/Cu-HNG 中的Fe已氧化。与Fe-HNG相比,Fe/Cu-HNG中的Fe K边略微向高能量移动,这意味着HNG中的Fe/Cu二聚体由于Cu配体的存在而略微提高了Fe的氧化态。与Cu-HNG 相比,Fe/Cu-HNG中的铁原子将电子转移到Cu上,使Cu的氧化态略有降低。吸收光谱得出的这种趋势与XPS分析结果一致。铜 K边EXAFS光谱显示,Fe/Cu-HNG、Cu-HNG 和Cu/CuHNG的主要峰值位于~1.45 Å处,对应于Cu-N配位的第一层配位。Fe-HNG 和Cu-HNG 的第二个峰分别位于2.15和2.27 Å处的第二个峰与铜箔的第一壳距离(2.24 Å)相当,表明存在金属-金属二原子构型。同样,Fe/Cu-HNG、Fe-HNG和Fe/Fe-HNG在~1.48 Å处的主要峰归因于Fe-N配位。图 3f 中 Fe/Cu-HNG、Fe-HNG 和 Fe/Fe-HNG 在 2.15 和 2.03 Å 处的第二个峰,进一步证实了金属-金属二原子构型的存在。k3 -加权傅立叶变换光谱表明,Fe/Cu-HNG中的金属间距短于Cu/Cu-HNG中的Cu-Cu 配位,长于Fe/Fe-HNG中的Fe-Fe配位,从而验证了Fe/Cu-HNG中存在异质的 Fe-Cu位点。图 3g-j 显示了小波变换(WT)的 EXAFS 光谱。因此,WT和FT-EXAFS分析证实了Fe/Cu-HNG 中存在金属-N 配位和金属-金属键的存在。实验结果与拟合结果的良好一致性证实了铁/铜双原子锚定在MN2位上,相邻的铁/铜原子结合在一起形成金属-金属二聚体结构。
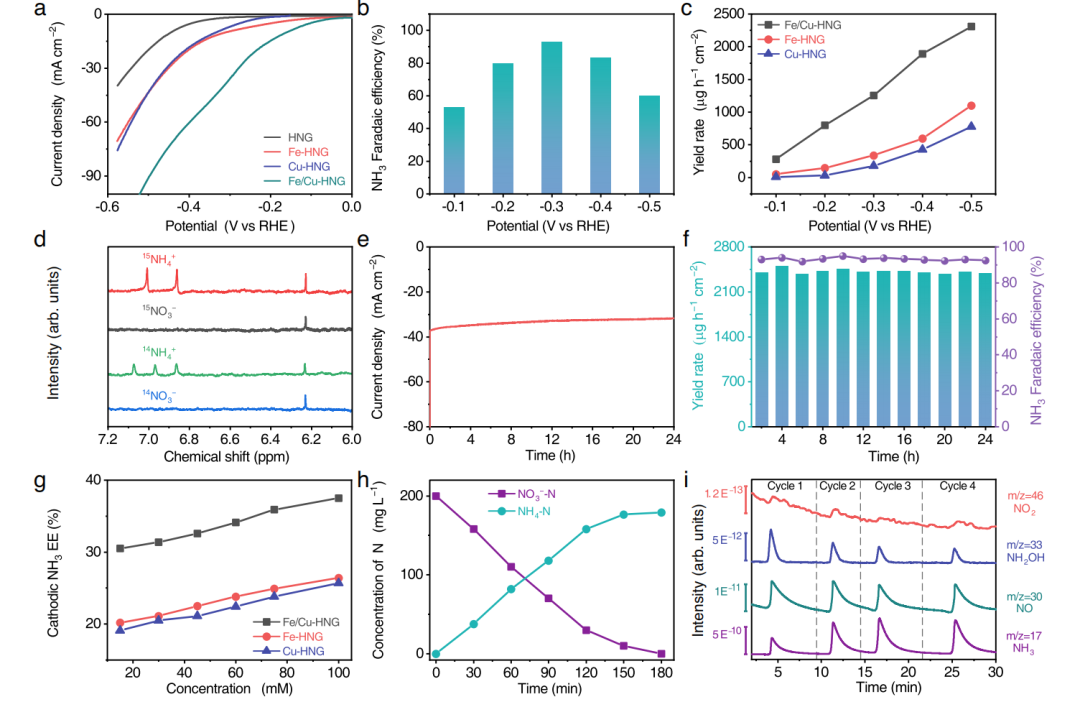
图四:NO3−RR的电催化性能
LSV曲线显示杂原子双位点催化剂(Fe/Cu-HNG)显著提高了电流密度。图4b显示Fe/Cu-HNG在- 0.3 V的电压下具有更高的FENH3最大值(92.51%),表明双原子催化剂比单原子催化剂具有更高的催化活和选择性。同位素标记法证实产物NH3实际上来源于NO3-RR而不是污染物。延长反应时间和催化剂回收实验表明Fe/Cu-HNG催化剂具有电化学稳定性。图4g显示了不同浓度下NH3生产的能量效率(EE)。由于过电位较低,Fe/Cu-HNG表现出比Fe-HNG和Cu-HNG高得多的EEs。对反应池中N的种类测试表明NO3−转化为NH3。在线差分质谱(DEMS)证明中间体NO2、NO、NH2OH、及主要产物NH3。
NO3−RR机理的理论分析
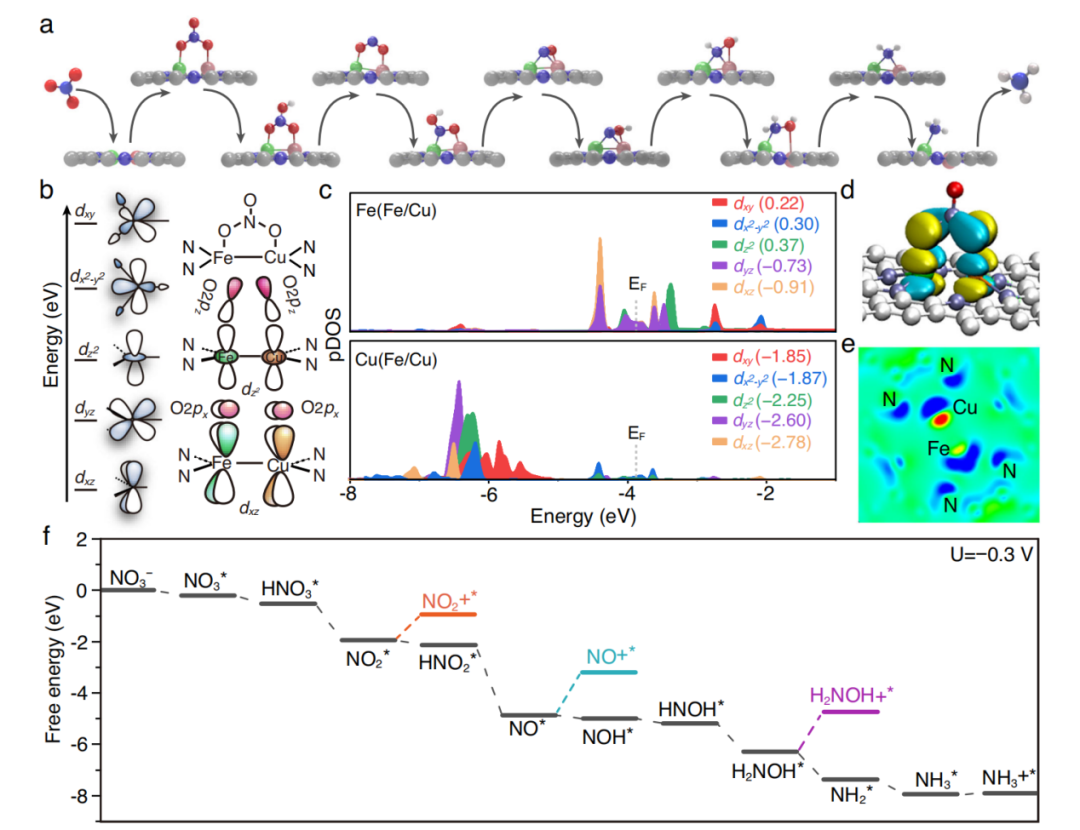
图五:NO3−RR机理分析
首先根据DEMS证明的NO2、NO和NH2OH的信号和强度预最可能的反应途径。在图5b中,Fe/Cu-HNGFe三维轨道的能级分裂及其与NO3−的相互作用。图5c给出了计算得到的Fe/Cu-HNG d轨道的偏态密度(PDOS)。通过对这些三维轨道的积分,本文得到了每个d轨道的相对能级。它们的相对位置与图5b中的MO理论分析一致。此外,当电压极化到−0.3 V vs RHE时(图5f),所有的台阶都是下坡的,NO3−RR自发发生。因此,Fe/ Cu-HNG的双原子位置适当地协调了初始放电和后续加氢/脱水步骤对降低能垒的冲突要求,从而显著提高了NO3−RR从NO3−到NH3的催化活性。
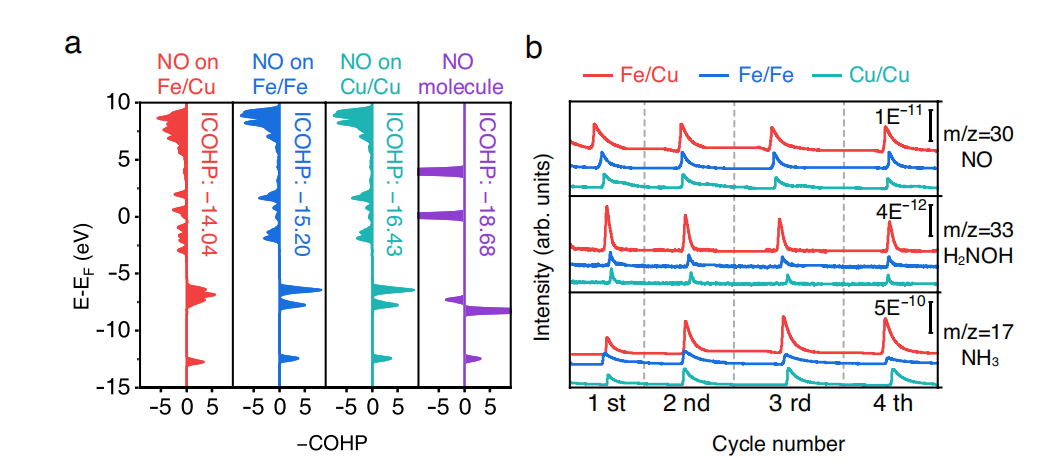
图六:NO3−RR中间体8NO活化机理分析
为了进一步阐明随后的脱氧/氢化反应,本文计算了吸附在Fe/Cu、Fe/Fe和Cu/Cu双原子位上的NO分子的晶体轨道汉密尔顿族(COHP)。图6a中的综合COHP (ICOHP)可以作为N-O活化的定量指标。NO分子在Fe/Cu位点上的ICOHP相对较高(−14.04 eV),表明由于异原子结构,N-O键明显减弱。活化的N-O键将促进随后的氢化。从图6b可以看出,Fe/Cu与Cu/Cu(或Fe/Fe)的NO产率比在2左右。由于Fe/Cu位点的显著活化,Fe/Cu-HNG的NH2OH/NH4产率显著提高,比Cu/Cu或Fe/Fe催化剂高8-10倍。还应注意的是,NH3信号的强度尺度比中间体高两个数量级。这些实验和理论结果证实,Fe/Cu-HNG可以降低NO3−RR的能垒,激活NO分子,从而提高NH3的收率和选择性。
结论:本文制备Fe/Cu双原子催化剂,在-0.3 V vs RHE条件下具有~38.5 mA cm-2的高活性和92.51%的FE还原NO3−为NH3的选择性。在-0.5 V条件下,NH3的产率达到1.08 mmol h−1mg−1。Operando DEMS和DFT计算揭示了NO3−到NH3的反应途径和转化机理。与Fe/Fe和Cu/Cu构型相比,Fe/Cu双原子位为大多数中间体提供了介质相互作用,降低了NO3−转化为NH3的总能垒。
论文相关信息
第一作者:Shuo Zhang
通讯作者:Qingshan Zhu, Huigang Zhang, Jun Lu
通讯单位:State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China,School of Chemical Engineering, University of the Chinese Academy of Sciences, No. 19(A) Yuquan Road, Shijingshan District, Beijing 100049, PR China,College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, Zhejiang Province 310027, China
*本文系转载,如涉及版权等问题,请联系我们以便处理
 电话:400-9933-062
电话:400-9933-062 电子邮箱:business@wykt.com
电子邮箱:business@wykt.com

