【文献解读】细菌病原体对宿主细胞途径的操纵

传染病是全世界人类健康的主要威胁,人们作出了巨大努力来了解各种传染因子及其毒性机制。在对细菌病原体的早期研究中出现的一个主题是,许多病原体将致病因子(也称为效应因子或毒力因子)直接注入宿主细胞,作为其致病策略的一部分,这些致病因子专门针对宿主的关键细胞内通路。这种致病性策略被某些细胞外和细胞内的病原体所使用。
细菌感染的细胞生物学
细菌病原体使用一系列效应物来破坏和控制正常的细胞功能。效应器通常是由Ⅲ型分泌系统(T3SS)或Ⅳ型分泌系统(T4SS)直接注入到宿主细胞的细胞质中的特化蛋白。这些分泌系统由一个结构保守的蛋白质器官组成,形状像针。
分泌蛋白作为微生物毒性剂的功能并不是新的概念。几十年来,人们已经认识到毒素具有这种能力。但是,在考虑感染过程的细胞生物学时,直接将效应器注入哺乳动物或植物细胞的能力是反复遇到的。因此,它是目前所理解的微生物发病机理中的一个重要过程,是病原体与宿主之间的重要界面。
为了促进它们的生命周期,细菌病原体可以利用宿主细胞来帮助它们自身的黏附、复制和/或传播。细菌定植的第一步是黏附。细菌病原体有多种细胞表面黏附素,包括菌毛黏附素(fimbriaeadhesins)和非菌毛黏附素(afimbrial adhesins),使它们能够附着在宿主细胞上。其中一些黏附素还有一个更深远的作用:它们与非吞噬细胞上的同源受体结合,从而使细菌被这些细胞吸收。这种具有双重作用的粘连蛋白包括耶尔森氏菌的侵入蛋白和李斯特菌的内部蛋白。细胞内病原体的内化机制因作用因子的不同而不同,它们需要占用补充的宿主细胞内信号通路。
病原体最常描述的细胞靶点是细胞骨架。各种细胞内微生物利用细胞骨架成分进入宿主细胞,并在宿主细胞内推动自己(图1)。细菌病原体通常不直接与肌动蛋白丝本身相互作用。相反,它们通过传递效应器的作用,通过调节细胞调节因子,如Rho样小G蛋白5,来破坏和控制肌动蛋白丝的聚合(图1a)。
一些细菌病原体在内化后仍留在液泡中,这些微生物通常使用效应器来调节泡内运输,在宿主细胞内提供一个保护位点(图1c)(包括在通常杀死细菌的巨噬细胞和中性粒细胞中)。此外,病原体可与细胞死亡途径(包括凋亡)相互作用,调节宿主-细胞死亡,促进病原体在宿主中的存活。此外,病原体规避或破坏宿主免疫反应(包括先天免疫和适应性免疫机制)的关键方法之一是分泌效应蛋白(图1d)。这些定植和免疫逃逸过程的步骤将在后面更详细地讨论。
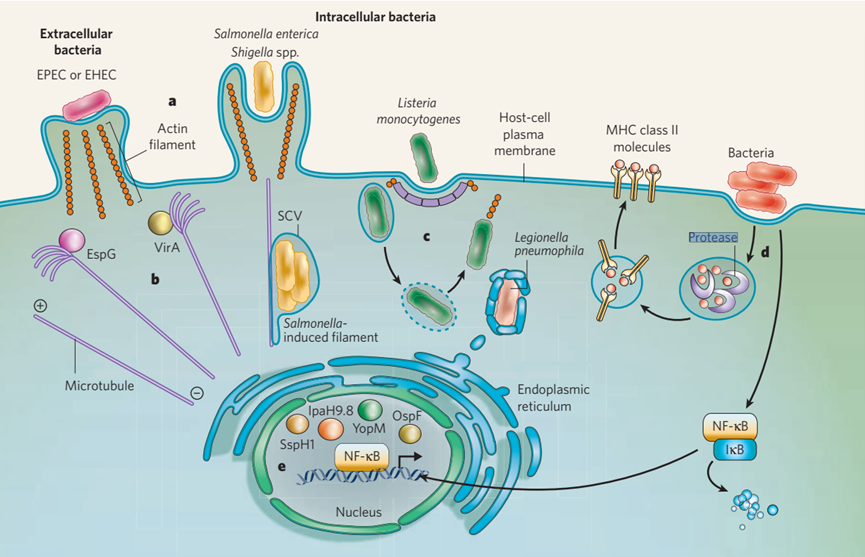
图1细菌感染的细胞生物学。胞外细菌病原体和胞内细菌病原体最初都与宿主细胞的质膜相互作用。这些病原体占据了各种常见的结构和途径。a,细胞外病原体(例如EPEC和EHEC)通常操纵肌动蛋白的细胞骨架,产生富含肌动蛋白的支架结构。相比之下,侵入性细胞内病原体肠道沙门氏菌和志贺氏菌利用这种细胞骨架成分入侵宿主细胞。另一种侵入性细胞内病原体,单核增生李斯特菌,利用网格蛋白介导的内吞作用进行入侵。b,在某些细菌感染过程中,微管,细胞骨架的另一个组成部分,通过特定效应蛋白的作用被分解。在EPEC感染(EspG)和福氏志贺氏菌感染(VirA)中存在的这些蛋白质会分解细菌接触附近的微管。c,侵入性细菌进入宿主细胞后,它们占据一个液泡。液泡可以提供对免疫检测的保护,并可以是一个复制位点(replicative niche)(在肠链球菌的情况下)。或者,细菌可以逃离液泡,获得推动自己通过细胞质的能力(在单核增生乳杆菌和福氏乳杆菌的情况下)。细胞器组分也可获得:例如,嗜肺军团菌通常通过吞噬作用内化,与内质微网衍生的囊泡相互作用。d,细菌病原体也有其他以效应蛋白为基础的机制来规避先天免疫反应和适应性免疫反应。例子包括颠覆细菌抗原在细胞表面的呈递(适应性)和干扰促炎转录激活因子NF-κB到细胞核的易位(先天性)。e,细胞核也是几种T3SS效应器的目的地(例如,小肠链球菌SspH1,耶尔森菌YopM,链球菌OspF和IpaH9.8),其影响开始显现出来。
操纵细胞骨架和膜结构
本节将讨论致病菌如何利用宿主细胞的细胞骨架、膜结构和关键信号通路,以及微生物入侵、在细胞内存活和在宿主体内复制的策略和原理。
细菌与肌动蛋白细胞骨架的相互作用
正如前面提到的,细菌病原体操纵细胞骨架来帮助入侵宿主细胞和/或获得细胞内的运动能力。它们经常与肌动蛋白丝相互作用,特别是通过调节G蛋白。侵袭性细菌肠道沙门氏菌与哺乳动物细胞的相互作用就是这一过程的例证。在这个过程中,肠球菌将T3SS效应蛋白SopE和SopE2传递到宿主细胞。这些效应物作为G蛋白的鸟嘌呤核苷酸交换因子,激活G蛋白CDC42和靶细胞中的G蛋白RAC家族。这种G蛋白的激活,反过来又诱导产生富含肌动蛋白的膜褶皱,从而吞噬和内化细菌(图1a)。
在侵入并从膜包裹的囊泡逃逸到细胞质后,许多病原体还操纵肌动蛋白-丝的动力学,以便它们能在受感染的宿主细胞内移动。通过细菌蛋白介导的肌动蛋白成核,它们将肌动蛋白招募到它们的一个极点。例如,福氏志贺氏菌的细胞内运动是由细菌效应剂IcsA介导的。IcsA与宿主蛋白N-WASP(神经Wiskott-Aldrich综合征蛋白;也被称为WASL),它反过来招募一个被称为ARP2 /3复合体(由七个宿主蛋白质组成,包括肌动蛋白相关蛋白2 (ARP2)和ARP3)。这种复合物聚合了前进细菌背后的肌动蛋白丝。相比之下,李斯特菌的胞质活性是由细菌蛋白ActA介导的,它直接与Arp2/3复合物和肌动蛋白相关蛋白VASP(血管扩张剂刺激的磷蛋白)结合。
细胞外病原体感染时,肌动蛋白相关的细胞骨架成分也会发生劫持。例如,附着和去除人类病原体肠出血性大肠杆菌(EHEC)和肠致病性大肠杆菌(EPEC)有一个复杂的细胞吸收过程(图2)。在这种情况下,细菌效应蛋白Tir介导黏附微生物下宿主细胞肌动蛋白丝的广泛修饰。Tir通过T3SS进入靶细胞,并嵌入质膜中,通过与细菌外膜蛋白内膜结合,牢牢固定。在EPEC感染的情况下,Tir在宿主细胞质膜的细胞质表面被酪氨酸磷酸化,并招募宿主适配器蛋白Nck(酪氨酸激酶的非催化区域)。这导致在宿主细胞表面形成一个“基座”,病原体就居住在那里。这些基座可以在细胞表面移动(“滑行”):基座中已经发现了肌动蛋白分解蛋白丝切蛋白和凝溶胶蛋白,这些蛋白质可能与肌动蛋白组装蛋白一起调节肌动蛋白丝的动态。
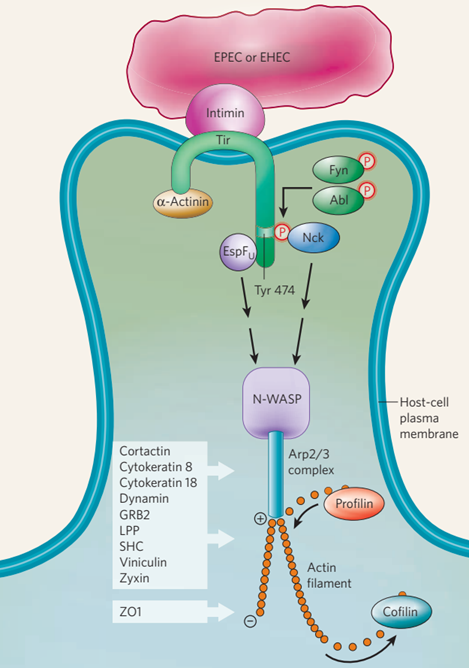
图2用EPEC和EHEC生成基座。在细胞外细菌EPEC的感染过程中,内膜受体(Tir)进入宿主细胞并插入宿主细胞的质膜(这一过程由T3SS介导)。这种受体在细菌表面与内膜相互作用,从而坚定地将细菌固定在宿主细胞上。EPEC Tir的羧基末端在474位酪氨酸残基上被至少两个宿主蛋白激酶Fyn和Abl磷酸化,导致宿主适配器蛋白Nck被吸收并直接与Tir结合。相比之下,EHEC感染期间,酪氨酸磷酸化事件被EHEC效应子EspFu破坏,因此不需要Nck。在EPEC或EHEC感染期间,N-WASP和Arp2/3复合体(由7个宿主蛋白组成)在tir相互作用蛋白(Nck或EspFu)的下游被募集,导致附着的细菌下方产生肌动蛋白丝,并形成基座结构。
微管与效应器的相互作用
微管通常也是微生物的目标。在微管相关分子马达蛋白的帮助下,这些极化结构通常用于结构支撑和作为引导和运输细胞内货物的轨道。在某些感染过程中,病原体可以改变和控制货物运输和微管组装和/或拆卸动力学。例如,在志贺氏菌的入侵下,VirA蛋白直接与α-微管蛋白和β-微管蛋白的异二聚体相互作用,促进微管的不稳定(图1b)。这导致入侵细菌附近的局部微管缺失,从而帮助志贺氏菌的入侵。在EPEC感染中可以看到类似的表型(图1b)。在这种情况下,局部微管解聚依赖于细菌效应子EspG,它与VirA类似,直接与微管蛋白相互作用。志贺氏菌和致病性大肠杆菌可以分解微管,而空肠弯曲杆菌的一种菌株已经被证明可以利用微管及其相关的分子马达来帮助入侵。
寄主细胞中的细菌生活
侵入性病原体进入宿主细胞后,要么局限在细胞质中,要么隔离在囊泡结构中。据推测,所有的细胞内病原体在其细胞内阶段的某一时刻占据一个膜包围的隔间,即使只是短暂的。内化后的初始间室(液泡和修饰后的吞噬体)由膜质宿主细胞组成;因此,内部化通常会产生保护区。病原体在细胞质中存活并散布到宿主体内之前,会采取各种策略在这些结构中繁殖或逃离。
占据受保护的细胞内位点的能力促成了肠链球菌和军团菌的发病。在吞噬细胞的被动内化过程中,军团菌占据一个称为含军团菌液泡(LCV)的腔室。这个吞噬体被Dot/Icm分泌系统(T4SS)修饰。T4SS效应物使LCVs既能避开常见的吞噬降解途径(通过防止液泡酸化和晚期核内体和溶酶体中发现的蛋白质与LCVs的结合),又能获得分泌途径中常见的成分。军团菌通过使用T4SS效应子SidJ进一步修饰吞噬体,将小的内质网衍生的囊泡招募到吞噬体膜上,然后介导与这些囊泡的融合,这可能为细菌提供丰富的营养资源(Fig. 1c)。此外,在LCV膜上发现了GTPase RAB1,并在内质网衍生的囊泡与LCV的融合中发挥作用。RAB1的功能由细菌效应器DrrA(也称为SidM)控制,随着感染的发展,这些囊泡消失(根据形态特征进行评估),发现核糖酶与LCV膜进行作用,从而将细菌放置在一个粗糙的、像皮肤状的液泡中。
同样,肠球菌通过使用一组T3SS效应剂改变其吞噬体样液泡,提供细菌生存和复制的保护位点(图1c)。它通过选择性地与宿主细胞内吞机制的组成部分相互作用来实现这一点,从而获得早期核内体抗原1和溶酶体相关膜蛋白1等分子。尽管溶酶体与含沙门氏菌液泡(SCVs)融合被抑制的观点早已被接受,但最近的进展表明,溶酶体在肠链球菌感染过程中可以很容易地与SCVs融合3,这就提出了许多关于逃避宿主破坏的确切机制的问题。
福氏志贺菌的主动入侵在入侵微生物周围产生液泡。然而,在上皮细胞侵袭过程中,福氏志贺菌只短暂地占据这个液泡。这种逃离液泡的过程允许细菌在宿主细胞的细胞质中复制,并最终从一个细胞传播到另一个细胞。单核增生李斯特菌是一种通过网格蛋白介导的内吞作用内化的病原体,最初也在液泡中发现(图1c)。类似于福氏志贺菌的入侵方式,这些液泡的寿命很短。单核增生李斯特菌利用膜孔毒素李斯特溶菌素O以及酶PlcA和PlcB破坏周围的膜,从而使其从液泡中逃逸,随后在宿主细胞胞浆内复制,并通过肌动蛋白介导在细胞间传播。
病原体保存
很明显,细菌病原体使用不同的机制来实现一个相似的目标——与宿主细胞相互作用,并可能改变宿主细胞。然而,细菌病原体也必须确保它们自己在宿主中的保存:它们需要避开免疫反应,这有利于复制和传播,这是任何病原体成功的必要条件。
炎症与转录因子NF-κB
先天免疫的基础是转录因子NF-κB基因的表达(图3)。这一过程是在细菌病原体相关分子模式(PAMPs)被模式识别受体(PRRs)检测到后诱导的,包括Toll样受体和NOD(核苷酸脱脂寡聚结构域蛋白)样受体(NLRs)。NF-κ b响应基因包括那些编码促炎细胞因子、抗凋亡因子(如Bcl-2)和防御素(一类抗菌肽)的基因。在这些基因被转录之前,NF-κB需要被激活,当它的细胞质结合伴侣IκB抑制剂被降解时,NF-κB就会被激活,从而使NF-κB能够转移到细胞核。NF-κ b响应基因包括那些编码促炎细胞因子、抗凋亡因子(如Bcl-2)和防御素(一类抗菌肽)的基因。IκB蛋白被蛋白IκB激酶磷酸化(IKK)后发生降解,它的活性是由PRRs刺激的。磷酸化的IκB被泛素修饰并进行蛋白水解降解。
病原微生物已经开始“了解”NF-κ b激活途径,并发展出规避它的策略(图3)。例如,福氏志贺菌和耶尔森氏菌都可以阻止IκB泛素化,从而阻止其降解,导致NF-κB在细胞质中保持活性。这些细菌通过T3SS效应蛋白OspG(来自福氏志贺菌)和YopP/J (YopP和YopJ是来自不同种类耶尔森氏菌的同源蛋白)起到作用。OspG与E2泛素结合酶UBCH5B(也称为UBE2D2)的泛素化形式结合,并阻止E3泛素蛋白连接酶将泛素转移到IκB,尽管如此IκB磷酸化作用仍在发生。相比之下,直到最近,已知YopP/J抑制NF-κB信号,但不清楚这是由于抑制IκB磷酸化还是由于IκB去泛素化。
有趣的是,福氏志贺菌通过在几个点上改变NF-κ b激活通路来避免先天免疫反应。最近的研究表明福氏志贺菌利用T3SS效应子OspF操纵编码NF-κ b响应基因的DNA的物理和空间环境48(图3)。通过DNA修饰(如甲基化)的表观遗传调控可对基因表达产生显著影响。OspF作为一个独特的磷酸酶。它在细胞核中去磷酸化有丝分裂原激活蛋白激酶(MAPK) ERK2,因此ERK2不能激活有丝分裂原和应激激活激酶1 (MSK1)和MSK2。这实际上阻止了组蛋白磷酸化,而组蛋白磷酸化是NF-κ b依赖性转录的先决条件。因此,通常被NF-κB激活的基因在检测到福氏志贺菌时保持沉默。
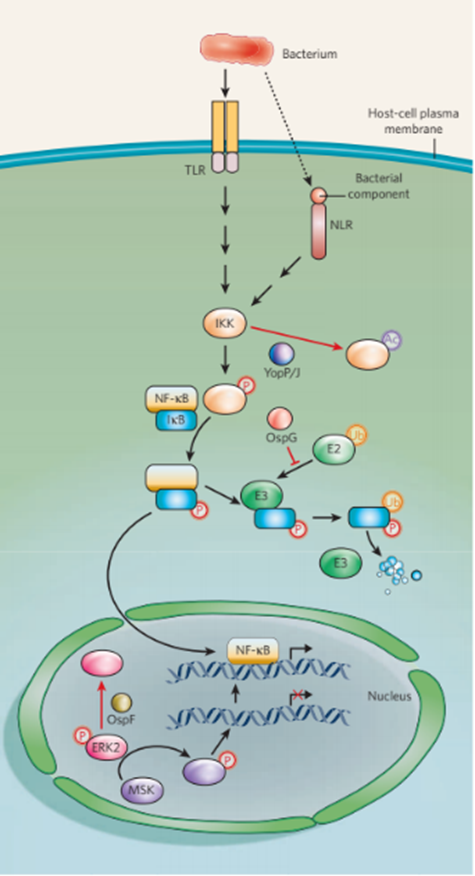
图3 NF-κ b介导的信号传导的破坏。转录因子NF-κB启动编码许多先天免疫因子的基因表达。因此,NF-κB信号通路对宿主至关重要。不同的病原体可以在不同的点上破坏这一途径。当致病菌被PRRs (TLRs和/或NLRs)检测到后,信号级联被触发,导致蛋白激酶复合物IKK的磷酸化。激活的IKK然后催化NF-κB抑制剂IκB的磷酸化。E2泛素结合酶携带的泛素,被E3泛素蛋白连接酶连接到磷酸化的IκB上,标记IκB在细胞质中降解,释放NF-κB转移到细胞核中。NF-κB诱导基因表达需要通过组蛋白的磷酸化重塑染色质。这是由激活的MSK介导的,MSK是一种被MAPK ERK2激活的蛋白激酶。致病菌分泌的蛋白效应子干扰NF-κB介导的基因表达的点用红色表示。YopP/J由耶尔森菌产生,通过竞争性乙酰化关键氨基酸残基来解除IKK的武装,从而阻止其磷酸化。福氏志贺氏菌在胞浆和细胞核两个点上破坏了这一信号通路。效应子OspG通过结合泛素化的E2分子“脱离”IκB泛素化。相比之下,另一种效应因子OspF通过去磷酸化激活的ERK来阻止染色质重构。
改变抗原表达
适应性免疫反应与先天免疫反应共同作用,但具有病原体特异性。因此,它最初需要精确的病原体识别,最终需要强有力的免疫诱导。细菌被称为抗原递呈细胞(APCs)的特化细胞识别和内化(图4)。然后微生物被隔离在特化膜包围的泡室中的蛋白酶降解。然后降解的微生物成分(多肽)与宿主蛋白质结合,称为主要组织相容性复合体(MHC) II类分子,这些抗原复合体被运输到宿主细胞表面,在那里它们被提交给其他免疫细胞。
几种微生物病原体破坏了适应性免疫反应的启动。虽然这些病原体使用的机制还没有在分子水平上被完全定义,但很明显,这些机制是多样的。例如,肠链球菌可以通过一种不完全确定的机制阻断树突状细胞(APC的重要类别)的抗原呈递(图4)。这是通过诱导受感染树突状细胞表面肽结合的MHC II类分子数量减少来实现的,从而激活(和增殖)较少的T细胞(适应性免疫细胞的重要类别)。
耶尔森菌也会改变树突状细胞的抗原呈递(图4),小肠结肠炎耶尔森菌使用T3SS效应子YopP/J,通过影响典型MAPK信号通路抑制适应性免疫反应。YopP/J抑制MAPKs JNK和p38 MAPK的磷酸化,导致树突状细胞通过网格蛋白介导的内吞作用吸收较少的抗原。据推测,MAPK信号级联中的这种干扰来自于YopP/J的乙酰转移酶活性,但这还有待确定。然而,通过减少抗原的吸收,耶尔森菌限制了T细胞的增殖,从而限制了适应性免疫反应。这些关于YopP/J具有不止一种效应的发现提出了一个有趣的想法,即效应蛋白可以在宿主细胞中具有多种功能。鉴于目前已确定的由细菌病原体产生的大量效应蛋白,多功能效应蛋白的存在是值得注意的。
耶尔森菌和其他微生物病原体在对抗适应性免疫反应方面有一个共同的高效策略。它们通过诱导细胞凋亡来根除APCs。YopP/J也是耶尔森菌感染过程中的一个关键成分。这种作用可能是由于NF-κB信号通路提供的抗凋亡信号刺激被取消。例如,NF-κB诱导抗凋亡调控因子Bcl-2的产生,如前所述,YopP/J可以有效破坏NF-κB信号。然后,当没有APCs存在激活T细胞时,这些免疫细胞的增殖被终止,从而停止适应性免疫反应。
有趣的是,志贺氏菌和肠球菌通过一种未知的NF-κb独立机制诱导巨噬细胞死亡,该机制依赖于通过关键的促炎激活因子caspase 1发出信号(图4)。最初人们认为这些细菌使用T3SS效应物诱导细胞死亡。然而,最近的证据表明,caspase 1的激活是通过IPAF(也称为NLRC4)检测细菌鞭毛蛋白介导的,IPAF是一种胞质PRR55。有趣的是,肠链球菌将鞭毛蛋白分泌到宿主细胞胞浆中依赖于T3SS的功能。caspase-1介导的巨噬细胞死亡(现在称为焦亡)的机制正在出现,但其存在的原因仍是个谜。事实上,诱导细胞死亡作为细菌免疫逃避策略是有争议的。然而,病原体诱导的宿主细胞死亡的结果与环境有关,因此可能对宿主有害也可能对宿主有益。
上面的例子说明了细菌病原体在与宿主细胞相互作用时,已经进化出不同的策略来确保它们的生存,这些最常见的策略包括通过分泌细菌效应器对宿主细胞内信号通路的操纵。考虑到特定信号通路中离散步骤的覆盖范围,微生物对真核生物信号通路的“理解”确实令人震惊。
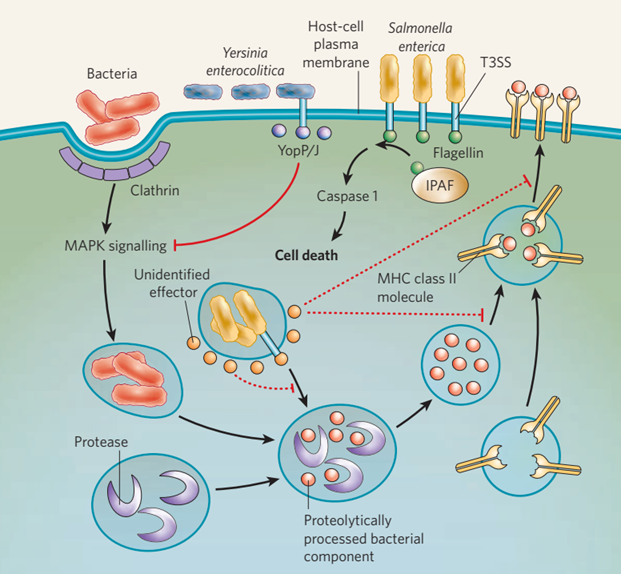
图4防止APCs呈递抗原。适应性免疫系统的组成部分在识别出现在受感染宿主细胞表面的病原体片段后被激活。致病菌的抗原以这种方式呈递:在细菌被带入内吞囊泡后,内吞囊泡与含有细胞蛋白酶的囊泡融合,在囊泡中细菌病原体被降解。经过蛋白质水解处理的细菌成分与MHC Ⅱ类分子结合,含有这些复合物的囊泡被运送到细胞表面,在那里细菌多肽展示给免疫细胞。细菌病原体在几个点上干扰这种抗原的处理和呈递途径,这些点用红色表示。小肠结肠炎耶尔森菌产生YopP/J,它通过抑制MAPK信号通路来破坏抗原呈递,从而阻止网格蛋白介导的内吞作用。肠道沙门氏菌分泌一种不确定的效应物来阻止抗原呈递,尽管目标步骤也不确定(虚线阻塞箭头)。此外,肠链球菌可通过诱导细胞死亡来阻止抗原呈递。这被认为是通过T3SS介导的鞭毛蛋白进入细胞质发生的。鞭毛蛋白随后被称为IPAF的PRR检测到,它启动信号级联,导致caspase 1的激活。Caspase 1通过新定义的称为焦亡的机制介导巨噬细胞死亡。志贺菌也被认为以同样的方式诱导巨噬细胞死亡。
未解决的问题
尽管在理解微生物发病机制的分子机制方面取得了进展,但对于在这一过程中起核心作用的细菌效应蛋白仍有许多有待了解。在这一节中,我们概述了一些关于效应器获得、进化和协调的重要未决问题。
毒力因子的选择和进化
很明显,微生物病原体具有与宿主细胞相互作用和操纵宿主细胞以及逃避宿主免疫反应的不同机制。据推测,细菌病原体进化出这些过程是因为它们提供了选择性优势。奇怪的是,病原体通常编码几个相关的效应器副本:例如,EHEC产生至少14个T3SS效应器NleG的变体,李斯特菌产生几个内部蛋白。尽管在某些情况下,这些蛋白质可能针对宿主细胞成分的相关变体,但在其他情况下,它们可能是多余的。尽管最近的证据表明,细菌效应蛋白对宿主之间的成功传播也至关重要,但为什么细菌病原体保持如此庞大的看似冗余的效应蛋白集合仍存在争议。
现在人们已经了解到,细菌之间(通过噬菌体、结合或转化)编码效应基因的遗传转移在产生病原体多样性(以及产生新疾病)方面起着关键作用。例如,最近有研究表明,细菌类核蛋白H-NS可以使基因在最初通过水平转移获得时沉默。然后,这些基因被整合到各种调控通路中,包括毒力调控通路(其中沉默可能被去除),允许编码的分子参与毒力调控通路
进攻的协调(Coordination of the attack)
毫无疑问,专门的分泌系统需要协调效应器的传递,以保持整体的毒力策略。理论上,病原体可以向不同类型的宿主细胞(例如,巨噬细胞和上皮细胞)或宿主的不同组织部位传递不同的专一效应器,并且不同的效应剂也可以用于针对不同的宿主物种。此外,可以想象的是,分泌系统可以反向工作:也就是说,它们可以为细菌获取宿主细胞分子、信号甚至能量和营养,尽管目前没有证据支持这一想法。最近的一项生物信息学研究表明,这些毒力系统的复杂性可能比人们想象的还要大。这项研究的作者提出,致病微生物可以通过一种称为末端重组的过程,立即产生新的T3SS效应子的嵌合杂交种。通过将新的蛋白质编码序列融合到控制T3SS效应子的表达和分泌的序列上,微生物可以对分泌效应子的新组合进行“采样”。这似乎加速了效应子进化,加上编码这些“打乱”效应子的基因倾向于定位于移动的遗传元件,并通过毒性或传播赋予强大的选择压力,表明致病微生物可以协调诱导的宿主细胞生物学,从而使发病机制优化为微生物的利益。宿主细胞中诱导的功能反应是如何协调的还不清楚
从更大的角度看发病机制
致病菌的艰巨任务是与宿主细胞相互作用,并重新编程这些细胞的复杂分子和细胞网络,以允许细菌复制和传播,同时对抗宿主的防御策略。通过(有时)大量编码效应蛋白的基因库的积累,进化和传播形成了细菌的这种追求,效应蛋白可能受到复杂的调控。零星研究可能有助于对其进行界定,但要更深入地了解其机制和潜在的干预点,最好是考虑到作为一个整体的提供、协调和机械功能。目前的挑战是集合跨学科工具箱(cross-disciplinary toolbox),使发病机制能够在“系统”水平上进行研究。
总结
细菌发病机理是一个迅速发展和发展的领域。随着大量效应蛋白功能的实现,整合众多宿主细胞靶标并将这一知识转化为对效应蛋白引起疾病的机制的准确理解是一个相当大的挑战。
认识到病原体可以通过使用无数的机制越过关键的宿主细胞通路,这使得人们对微生物学、细胞生物学、生物化学和免疫学有了更多的了解。然而,这方面的知识需要进一步发展,以便能够转化为对疾病的真正理解。这仍然是所有参与这一领域的人面临的重大挑战。只有这样,才有可能将这些效应机制作为一种预防或治疗策略,加以合理的定位。
参考资料
Bhavsar, A. P., Guttman, J. A., & Finlay, B. B. (2007).Manipulation of host-cell pathways by bacterial pathogens. Nature, 449(7164), 827–834.
doi:10.1038/nature06247
https://pubmed.ncbi.nlm.nih.gov/17943119/
*本文系转载,如涉及版权等问题,请联系我们以便处理
 电话:400-9933-062
电话:400-9933-062 电子邮箱:business@wykt.com
电子邮箱:business@wykt.com

