文献解读|(25.269):肝细胞色素P450 8B1和胆酸通过抑制肠道干细胞自我更新增强结肠炎肠上皮损伤
论文ID
原名:Hepatic cytochrome P450 8B1 and cholic acid potentiate intestinal epithelial injury in colitis by suppressing intestinal stem cell renewal
译名:肝细胞色素P450 8B1和胆酸通过抑制肠道干细胞自我更新增强结肠炎肠上皮损伤
影响因子:25.269
发表期刊:Cell stem cell
发表时间:2022.09.01
DOI号:10.1016/j.stem.2022.08.008
背 景
炎性肠病(IBD)是非特异性的慢性炎症性疾病,包括克罗恩病(CD)和溃疡性结肠炎(UC)等,目前尚没有非常合适的治疗方案。IBD的发病机制十分复杂,与宿主的肠道微生物、免疫系统、遗传组成等多种因素密切相关,因此深入了解IBD的致病因素迫在眉睫。胆汁酸(BAs)是胆固醇分解代谢的最终产物,作为两亲性乳化剂,能促进肠道脂质的吸收以及胆固醇和磷脂的胆汁排泄。BAs主要通过经典途径和替代途径在肝脏中合成。经典途径由CYP7A1起始,随后由胆固醇12α-羟化酶(CYP8B1)控制,该途径产生的BAs约占总胆汁酸的75%。CYP8B1是初级胆汁酸(CA)合成关键酶,它决定了非-12-OH BAs(CDCA,α/β-MCA,UDCA,LCA, HCA, HDCA及其他替代途径的衍生物)与12-OH BAs(CA,DCA)的比例。非-12-OH胆汁酸比例增加一般与代谢性疾病如肥胖呈正相关关系。除此之外,BAs的合成和代谢失调还与啮齿动物和人类的多种肠道炎症疾病有关,但BAs影响肠道炎症的机制目前尚不清晰。
实验设计

结 果
01
溃疡性结肠炎患者和结肠炎小鼠的胆汁酸代谢失调
作者首先分析了溃疡性结肠炎(UC)患者和健康人血清中各种胆汁酸的含量,结果发现 :UC患者血清中CA和DCA的含量以及12/非-12-OH胆汁酸的比例显著增加(图1A-1C)。并且,在DSS诱导的结肠炎小鼠模型中也能观察到这一现象(图1D-1I)。由于CYP8B1是促进CA合成的关键酶,决定了非12-OH BAs(如CDCA,α/β-MCA,UDCA,LCA, HCA, HDCA及其他替代途径的衍生物)和12-OH BAs(CA,DCA)的比例。因此作者推测这一现象与UC患者肝脏中CYP8B1表达异常增加有关,并在mRNA和蛋白水平上证实了这一猜测(图1L-1N)。
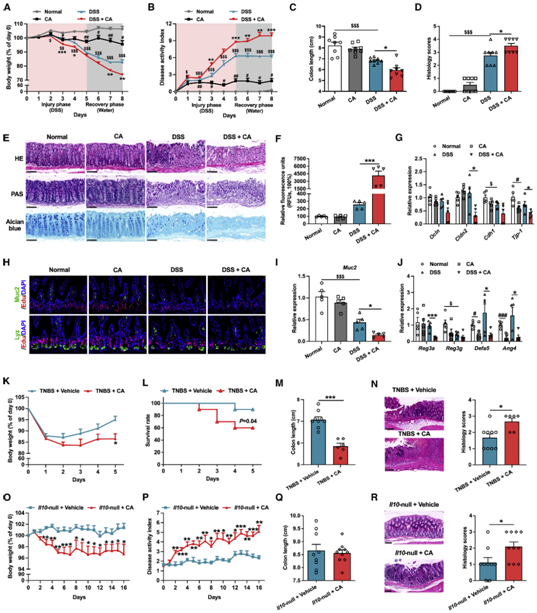
图1、UC患者和实验性结肠炎小鼠中cyp8b1-初级胆汁酸代谢途径被激活
02
补充CA会加重结肠炎小鼠的肠道损伤并加剧肠道屏障失调
随后,由于已有研究中没有直接证据表明CA会加剧结肠炎,为了研究CA与结肠炎之间的关系,作者向结肠炎小鼠补充CA后发现,CA在加剧DSS诱导的体重下降的同时,还导致肠道炎症的疾病活动指数(DAI)增加(图2A-2C),且加深了肠道粘膜屏障损伤(图2A-2E)。既往研究表明,粘膜屏障功能障碍有助于IBD的发展。为了进一步研究CA对肠道屏障的影响,作者随即利用异硫氰酸荧光素葡聚糖荧光探针( FITC-Dextran)检测CA对肠道屏障通透性以及破坏程度的影响,结果发现CA处理增加了结肠炎小鼠的肠道通透性(图2F)。
与之一致的是,CA还降低了结肠炎小鼠肠道中紧密连接蛋白2(Claudin 2)和黏蛋白2(mucin 2)的表达(图2G-2I),同时还降低了肠道中潘氏细胞的数量和抗菌肽的含量,这些结果表明CA破坏了肠道对病原微生物的防御机制(图2H, 2J)。另外,CA促进结肠炎小鼠肠道损伤的现象,在三硝基苯磺酸诱导的克罗恩病小鼠、IL-10敲除的自发性结肠炎小鼠中均得到了复制(图2K-2R)。但是在健康小鼠中,虽然CA单独处理即可诱导小鼠体重下降并导致DAI增加,却对小鼠的肠道形态、腹泻和便血情况以及肠道通透性没有显著影响(图2A-2J),说明CA本身并不能显著降低肠道屏障功能。
此外,虽然CA降低了结肠炎小鼠肠道中潘氏细胞的数量,但本身并不能在很大程度上影响IBD发病过程中的免疫反应(数据见原文S3A-3E)。以上结果表明CA在IBD发病机制中起着关键作用,病理水平的CA可与DSS诱导的肠道损伤协同增强肠道炎症,并主要是通过诱导肠道上皮粘膜屏障损伤而不是直接调节免疫细胞的免疫反应。
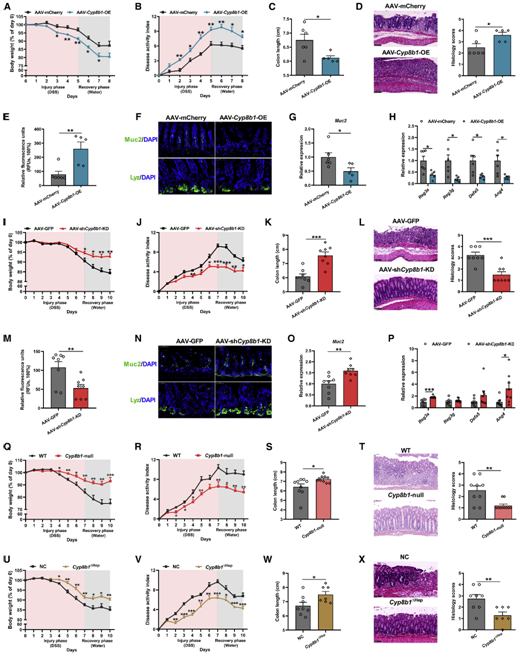
图2、结肠炎小鼠服用CA会加重肠道损伤
03
肝脏中特异性敲除CYP8B1能缓解肠道炎症水平
紧接着,为了研究CYP8B1在CA诱导的结肠炎小鼠肠道损伤中的作用,作者利用AAV腺病毒在小鼠肝脏中特异性过表达CYP8B1(AAV-Cyp8b1-OE)并进行DSS处理,结果发现CYP8B1过表达成功升高了CA水平,并产生了和外源补充CA的结肠炎小鼠相同的表型(图3A-3H),说明过度激活肝脏中的CYP8B1同样会引起上皮黏膜屏障功能障碍。反之,全身性或者肝脏特异性的抑制CYP8B1表达则能缓解结肠炎小鼠的肠道炎症(图3I-3X)。综上所述,肝脏CYP8B1可通过促进CA的合成来降低肠道屏障功能,进而加重肠道炎症情况。
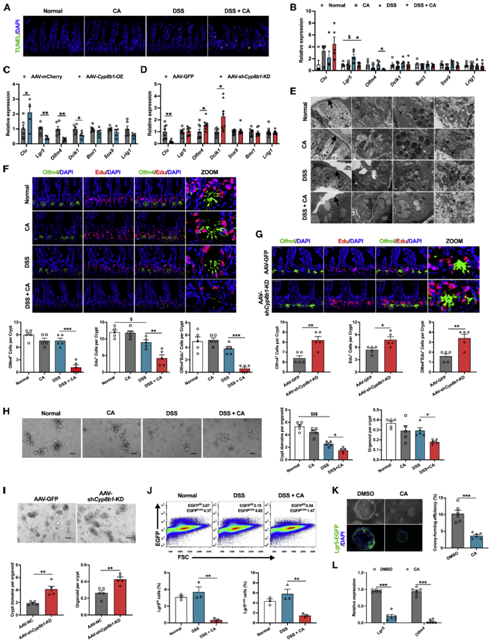
图3、肝脏过表达CYP8B1可增强结肠炎症,而CYP8B1敲除则可改善炎症
04
过量的CA导致Lgr5+的肠道肝细胞数量减少并抑制其自我更新
随后作者想研究的科学问题是,CA是如何影响肠道屏障完整性的?由于前期结果发现CA发挥作用并不依赖于肠道微生物,同时也不会对肠道免疫反应产生很大影响,于是,作者猜想CA是否是通过影响肠道中除免疫细胞外的其他细胞进而影响肠道屏障。
肠道屏障的构成较为复杂,外层包括粘液层、共生肠道微生物群和防御蛋白。肠上皮细胞(IECs)是中间层,在生理状态及病理状态下, 肠上皮细胞会不断的更新换代凋亡以帮助肠道的修复和再生,维持着肠道的正常结构和功能,对肠道屏障十分重要。作者便首先检测了CA对肠上皮细胞增殖和凋亡的影响。结果发现,单独的CA和DSS均不会显著促进肠上皮细胞死亡,但在 DSS 处理的同时补充 CA 却会抑制细胞的增殖,并损伤从隐窝底部到绒毛顶部在内的整个隐窝绒毛上皮部分(图4A和4B),且在AAV-Cyp8b1-OE结肠炎小鼠中也可以发现类似的结果,而Cyp8b1缺失后的表型却与之相反(图4C,4D)。作者进一步利用透射电子显微镜观察发现,CA处理的结肠炎小鼠的肠道上皮形态发现显著变化,这些变化包括杯状细胞丢失、潘氏细胞损伤、粘液和嗜酸性粒细胞胞质颗粒减少、线粒体肿胀等(图4E)。肠道上皮的更替由位于隐窝区底部的多能性LGR 5+ 隐窝基底柱状细胞(CBC)驱动。CBC包含三个亚群,通过分析CA处理和肝脏过表达CYP8B1对这三群细胞标志基因的影响,作者发现只有Lgr5+ intestinal stem cells(Lgr5+ ISCs)的数量显著减少,而抑制CYP8B1的表达则促进了IL-10敲除小鼠以及DSS诱导的结肠炎小鼠肠道中Lgr5+ ISC的细胞数量(图4B-4D,4F, 4G)。
肠道干细胞的分裂和分化是ISCs类群的基本特质,通过进一步对CA处理的结肠炎小鼠和肝脏CYP8B1过表达小鼠的肠道隐窝进行类器官培养,作者发现与对照组相比,上述两组的隐窝在体外生长分化速度更慢,且出芽率降低(图4H,4I)。进一步分析发现,CA处理改变了结肠炎小鼠隐窝中Lgr5+和Lgr5 -细胞的比例(图4J,4K, 4L)。这些观察结果表明,CA是通过抑制肠道损伤情况下Lgr5+肠道干细胞的自我更新来阻碍肠道的自我修复进而加重肠道损伤,最终导致肠道屏障损伤。
05
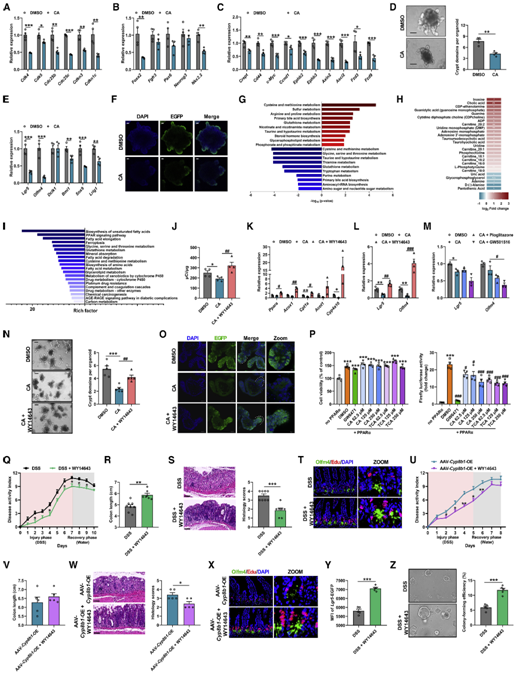
CA通过抑制脂肪酸氧化来损害Lgr5肠道干细胞自我更新能力
随即,作者首先在体外研究了CA是如何影响ISCs自我更新功能的。通过检测ISCs自我增殖及分化相关基因的表达,作者发现在CA处理后ISCs中与细胞周期、细胞分化以及Wnt信号通路相关的基因表达显著降低(图5A-5F)。由于近年来越来越多的证据表明细胞代谢深刻的影响着干细胞的增殖、多能性和分化。于是作者分析了CA对小鼠结肠隐窝代谢的影响,结果发现CA主要扰乱隐窝中的氨基酸和核酸代谢,这一结果与CA阻碍ISCs的细胞周期导致增殖能力降低相吻合(图5G)。CA同时还显著增加了隐窝中不同长度碳链的酰基肉碱水平,表明隐窝中的脂肪酸氧化受到抑制(图5H)。RNA-seq数据同样表明,CA显著抑制了隐窝中控制脂肪酸代谢的PPAR信号通路(图5I)。于是,作者进一步在体外检测了CA对隐窝脂肪酸β-氧化的影响,结果发现CA显著抑制脂肪酸的氧化能力并抑制了隐窝中与β-氧化相关基因的表达(图5J-5K)。以上结果表明,CA可能通过抑制PPAR信号通路以及脂肪酸氧化进而影响ISCs的增殖分化能力。
图5、CA治疗通过抑制Pparα信号传导以阻止Lgr5+肠道干细胞的脂肪酸氧化
06
CA通过抑制PPARα信号通路抑制肠道损伤修复过程
PPAR家族由Pparα,Pparβ和Pparγ三个成员组成。作者发现,只有利用激活剂WY14643特异性激活Pparα才能解除CA对Lgr5+ ISCs增殖的抑制,并促进隐窝类器官出芽。激活Pparα还减轻了DSS处理的小鼠以及Cyp8b1过表达小鼠的结肠炎症。作者进而通过肠道特异性敲除Pparα的结肠炎小鼠证实CYP8B1-CA代谢轴通过抑制Pparα介导的脂肪酸氧化,削弱Lgr5+ ISCs的更新能力,进而加重肠道屏障损伤(图6A-6C)。然而,外源性CA治疗既不会加重Pparα敲除小鼠的结肠炎症水平和肠道损伤,也不会影响ISCs的增殖能力(图6D和6E)。一致地,虽然源自Pparα敲除的结肠炎小鼠的隐窝,其出芽、类器官形成和自我更新能力存在缺陷,并且Lgr5+ ISCs标志基因的表达降低,但额外补充CA则对来自Pparα敲除的结肠炎小鼠隐窝没有显著影响(图6F-6I),并且,WY14643也不能促进Pparα敲除的结肠炎小鼠的隐窝类器官出芽(图6J和6K)。这些数据表明,CA是通过抑制Pparα介导的脂肪酸氧化损害Lgr5+ISC的再生功能,从而加剧结肠炎期间的肠道损伤。
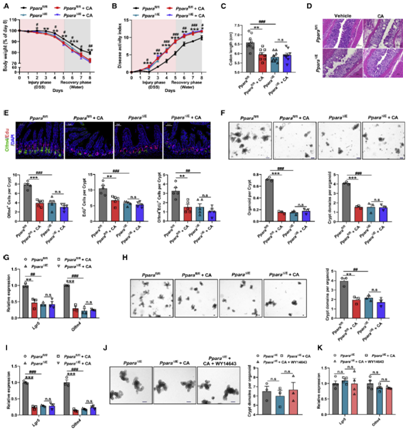
图6、CA对肠屏障功能的作用依赖于PPARα
07
OCA治疗通过靶向肝脏FXR-CYP8B1信号传导恢复受损的上皮粘膜屏障并缓解结肠炎
上述发现表明,CYP8B1可能是未来治疗IBD的潜在靶点。由于前人研究发现,肝脏或肠道的Farnesoid X受体(FXR)激活均可下调CYP8B1的表达。同时作者也发现,结肠炎小鼠中的肝脏和肠道中的FXR信号通路均受到抑制,于是作者进一步猜测激活FXR信号通路是否可以通过胆汁酸代谢缓解肠道炎症。值得注意的是,一种临床使用的FXR激动剂OCA,已在实验性的IBD模型中被报道可抑制肠道炎症并保护肠道屏障。然而,尚不清楚OCA对IBD的治疗作用是否涉及胆汁酸代谢的改变以及肠上皮的再生。与之前的研究一致,作者发现OCA能治疗DSS诱导的肠道炎症 (图7A-7J)。嗅觉调节素 4(Olfm4)是在发炎的结肠上皮细胞中选择性地表达的标志物,对Olfm4和Edu进行免疫荧光染色进一步证实,OCA促进了Lgr5+ ISC的增殖并抑制了肠道炎症 (图7K-7L)。与Pparα激动剂相似,OCA也增加了结肠炎小鼠肠道中Lgr5Hi细胞的数量,并维持了Lgr5Hi细胞在体外分化为类器官的潜力。(图7M-7N)。综上。这些数据表明通过激活FXR进而抑制肝脏 CYP8B1 的表达将是 未来IBD 的潜在治疗方案。
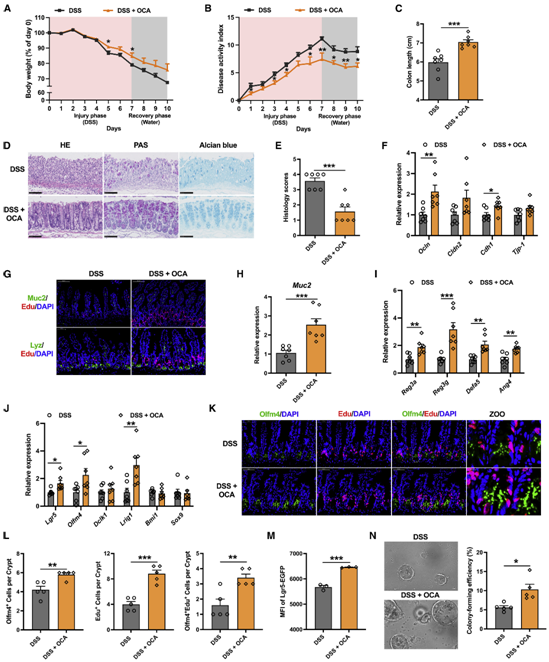
图7、OCA处理可通过改善上皮粘膜屏障功能缓解小鼠结肠炎
+++++++ ++ ++
结 论
本文发现,结肠炎期间肝脏CYP8B1的激活会导致CA在肠道中积聚,进而通过抑制Pparα介导的脂肪酸氧化导致Lgr5 +ISC功能障碍,并最终加重肠道损失。通过药理学特异性激活FXR或通过遗传手段特异性敲除CYP8B1则可抑制肠道中CA的含量并缓解结肠炎。
*本文系转载,如涉及版权等问题,请联系我们以便处理
 电话:400-9933-062
电话:400-9933-062 电子邮箱:business@wykt.com
电子邮箱:business@wykt.com

